Final project 4.1 Final Project
Project Title
| Structure Analysis | Mutation F19Y is found in Karl Zimmer’s genome for 4BMB (galectin-8). Analyze the structure of this mutation. |
Table of Contents
Introduction:
The gene 3VKO codes for the Galectin-8 protein, which functions as a Beta-galactoside-binding lectin. Galectin-8 is thought to act as a membrane damage sensor during infections, and restricts the proliferation of pathogens by targeting them for autophagy. Galectin-8 also detects endosomal membrane rupture events by binding beta-galactoside ligands on the lumenal side of endosome membranes, which are exposed to the cytoplasm following rupture of the endosome. The mutation of interest studied here is F19Y, a mutation of a phenylalanine to a tyrosine.
Writing:
From an initial side-chain chemistry standpoint, this mutation should be relatively minor, as it occurs away from the binding pocket, and the residue does not appear to play a major role in the interaction of the two domains of the protein. F19 can be found as part of a beta-barrel structure near the interface of the domains, but was not found to make any direct contacts in the interface, instead being part of a beta sheet. From a space-filling perspective, the tyrosine mutation is about as large as phenylalanine, and therefore may disrupt the structural stability of the protein, but not in any majorly appreciable way.
The F19I mutant, mutating the phenylalanine to an isoleucine, also has a minor role in the protein stability. The overall hydrophobic properties are similar, and so at worst the protein is destabilized, with the actual binding properties being minimally impacted. Similarly, F19L, mutating the phenylalanine to a leucine, should also have a minor role, as the hydrophobicity is not dramatically changed. Overall, there is more space in the structured regions by mutating from a phenylalanine to an isoleucine or a leucine, and so the structure of the protein and the flexibility of the beta-sheet is probably altered, but the actual protein should still have normal functionality and have minimal phenotypic effects.
Coding:
A) For the 4BMB F19 and F19Y residue, plot the total potential energy URLJ involving interactions among side chain atoms as a function of Chi_1 and Chi_2. Discuss how much you expect the wildtype protein to change to accommodate this mutation.
B) Calculate the root-mean-square deviation (rmsd) of the heavy atoms in the F19 and F19Y structures. How might this rmsd be related to the stability of the protein?
Documentation:
Codes, figures, and text files can be found in the StructAnal directory.
A) To generate heatmap plot of repulsive Lennard Jones potential energies, run:
python 4BMB_Urlj_Heatmap.py -u1 4BMB_F19.txt -u2 4BMB_F19Y.txt
To generate 3D plot of repulsive Lennard Jones potential energies, run:
python 4BMB_Urlj.py -u1 4BMB_F19.txt -u2 4BMB_F19Y.txt
Both programs make two-dimensional arrays of the χ1, χ2, and energy data to generate the plots.
B) To calculate RMSD between the WT and mutant 4BMB, run:
python 4BMB_RMSD.py -p1 4bmb_aligned.pdb -p2 4bme_aligned.pdb
Program extracts the relevant information from each PDB file, finds coordinates of backbone atoms C, N, O, and Cα for each residue in each protein, and calculates root-mean-square deviation for each residue. Spits out plot of residue vs. RMSD and total RMSD.
Results:
A) Phenylalanine and tyrosine are very similar in shape. The repulsive Lennard Jones energies associated with their rotations dip in very similar areas (χ1≈175°, χ2≈60°), suggesting that both are likely oriented in very similar positions with similar dihedral angles. The 3D plot shows direct comparison between the two residues (blue = F19, red = F19Y), where F19Y dihedral angle allowance, based on energy dip, is considerably narrower than for F19, perhaps due to the protruding -OH limiting movement. The heatmaps show more cleanly the allowed dihedral angles, and the heatmaps are not very different from one another. Thus, since F19 and F19Y are likely to be in approximately similar positions, we would expect that the rest of the protein will not deviate much from its original structure to accomodate the mutation.
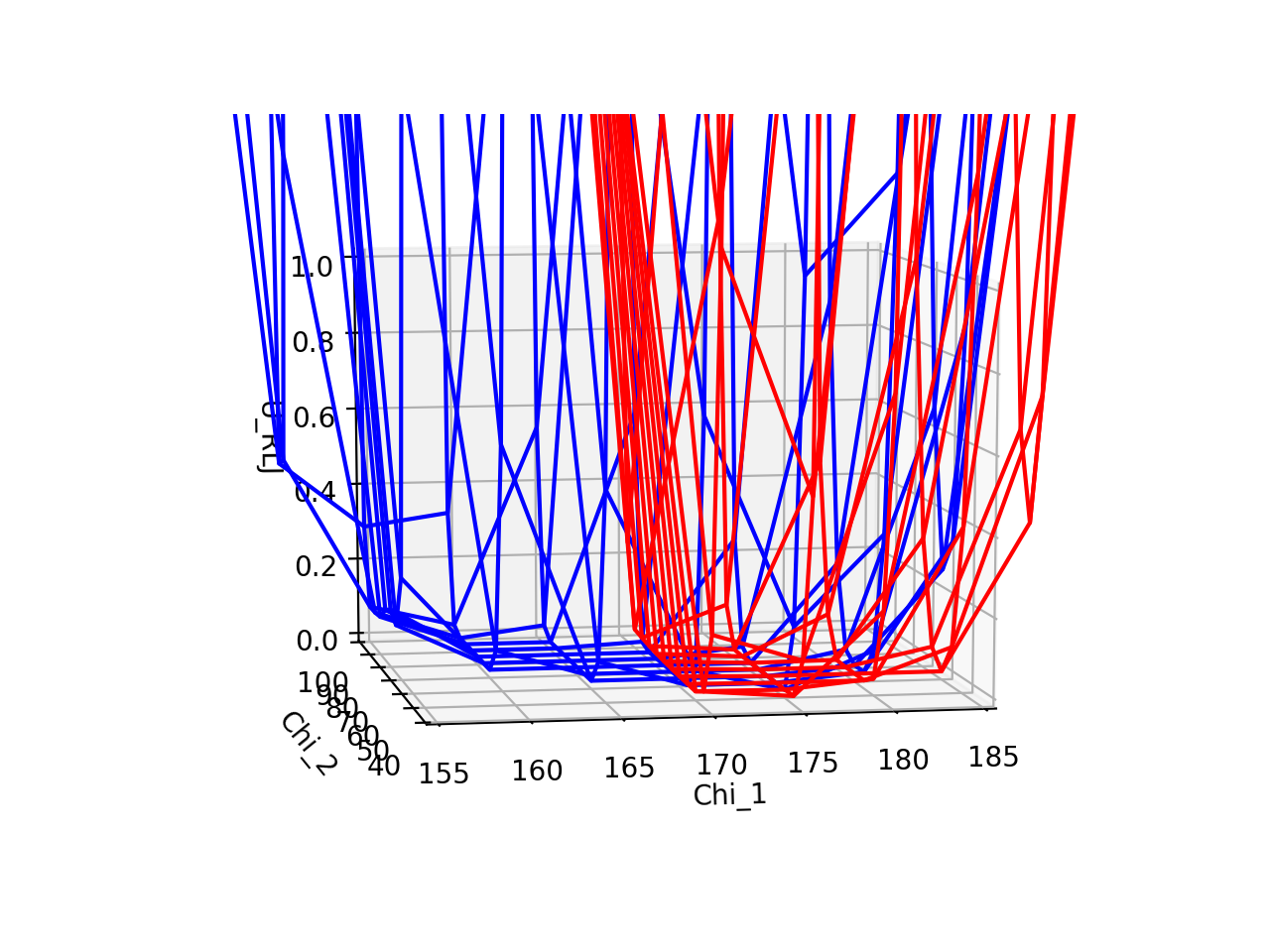
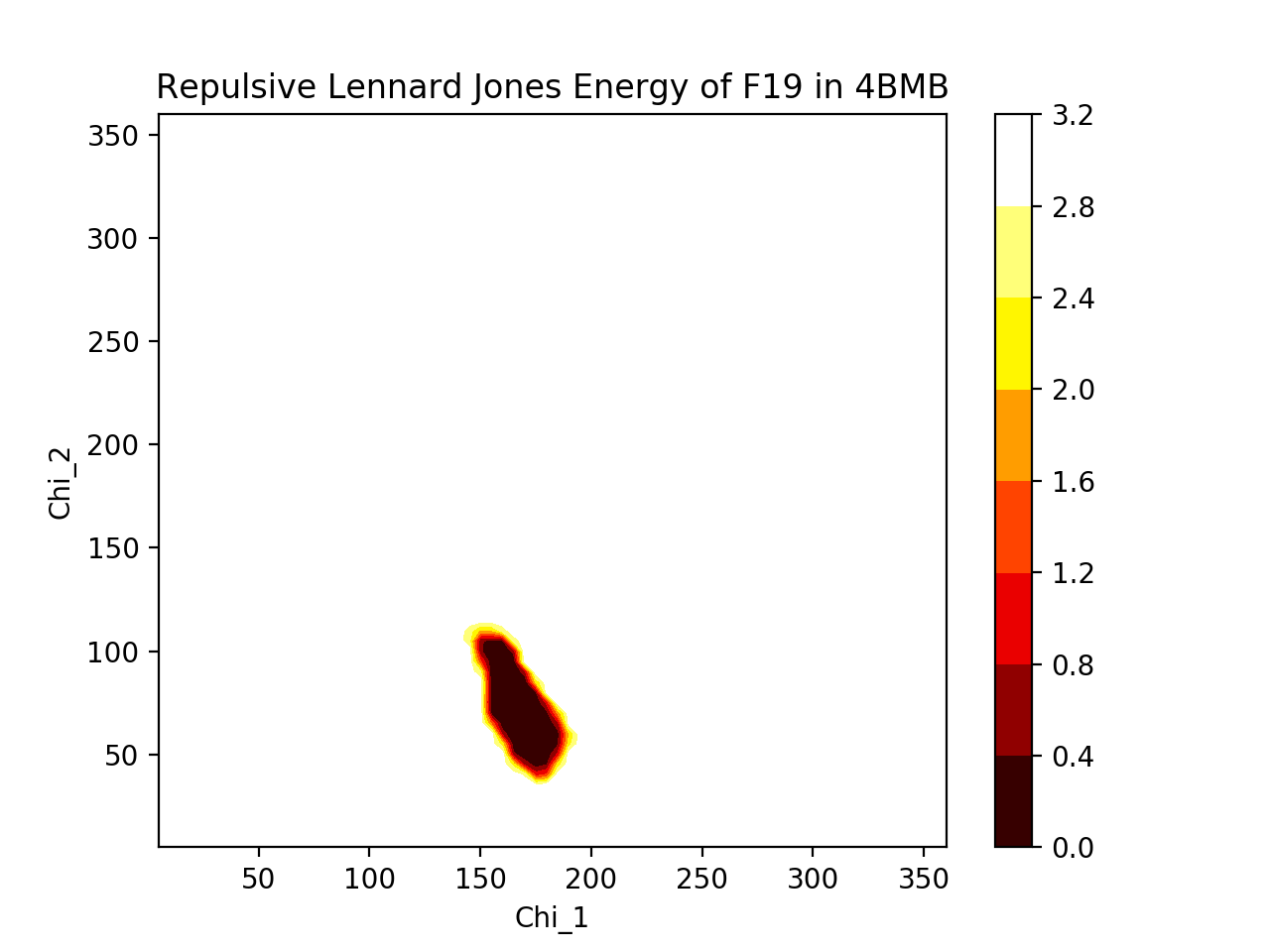
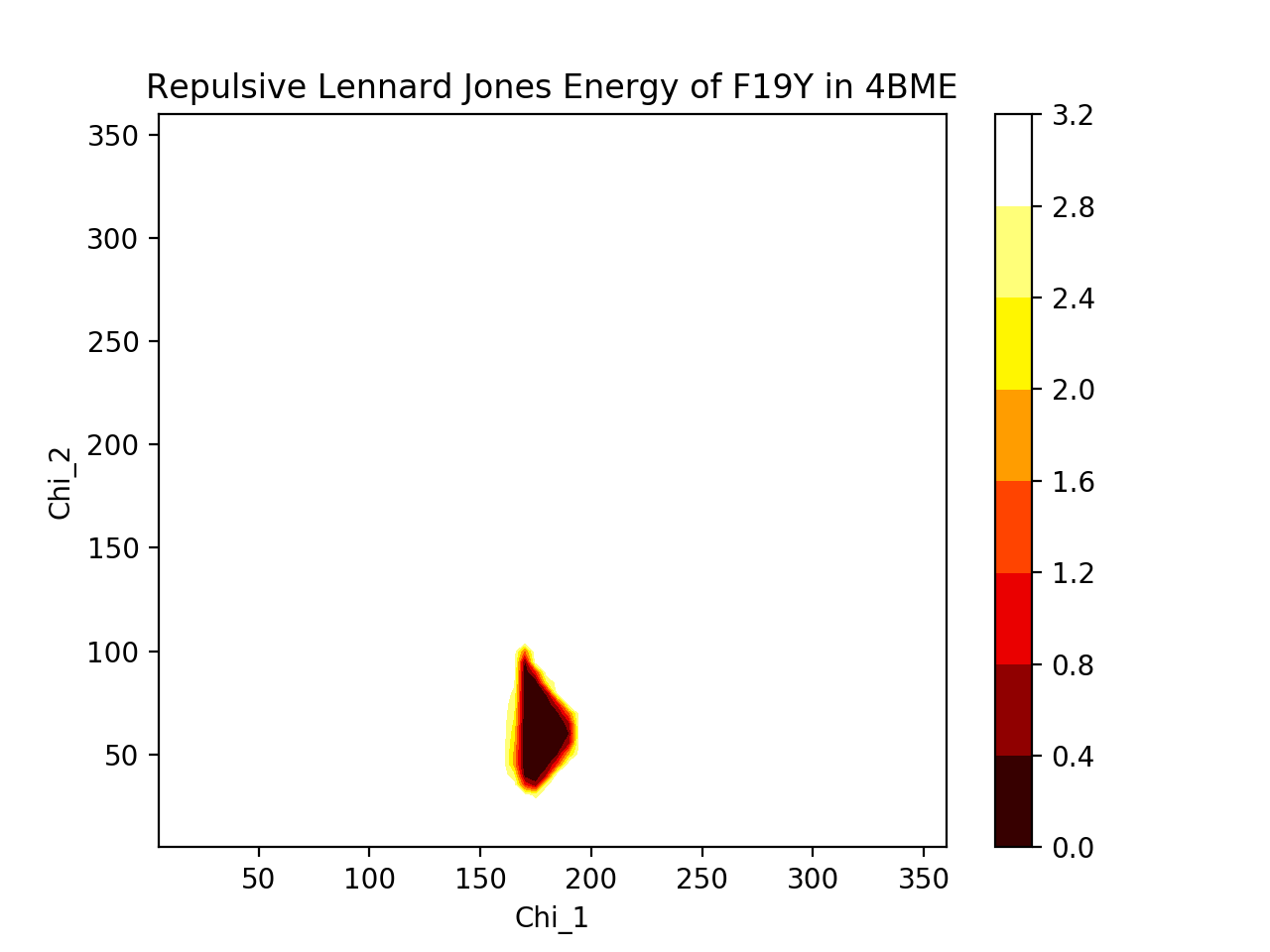
B) The root-mean-square deviations (RMSD) of backbone heavy atoms tell us how different the backbone positions are between two proteins. The total RMSD between 4BMB and 4BME is 0.3 Angstroms. Shown below is the RMSD for each residue. It is clear that there are fluctuations in the RMSD, and highest deviations occur at residues ~15, ~55, ~85, and especially at ~73. (Note that at residue 19, where the mutation occurs, RMSD is relatively low.) These fluctuations suggest that the protein is somewhat flexible and capable of some movement, and thus maybe a bit unstable (as we would expect less movement to correlate with stability, and more movement to correlate with instability). Despite this, the fluctuations are actually fairly small (apart from the one residue that deviates over an Angstrom), suggesting that, realistically, the mutation is probably not changing the protein drastically.
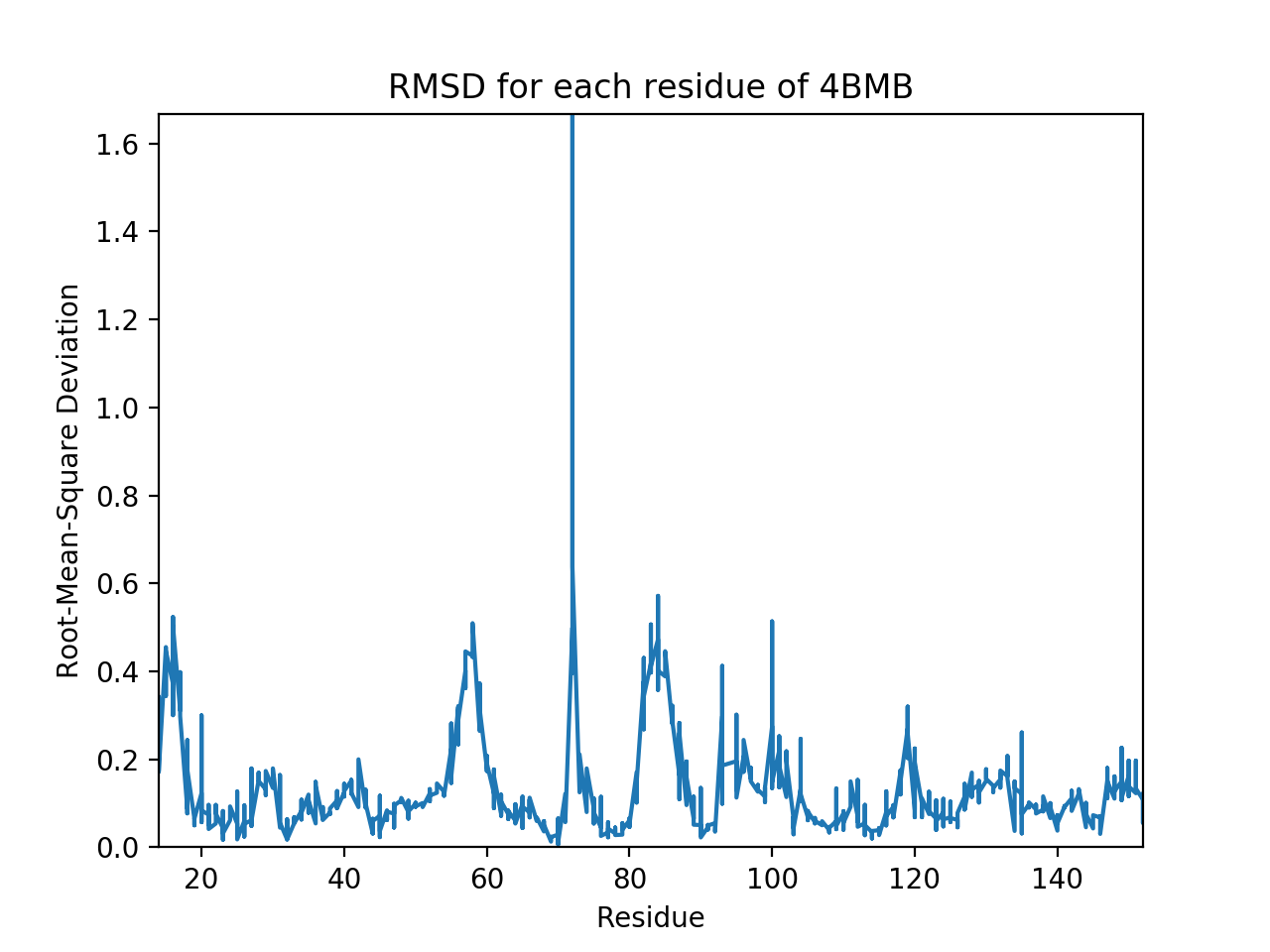
Pipeline:
Download the 4BMB structure from the Protein DataBank. Using the Rosetta Software Suite, generate 10 different protein mutations at location 19 (choose 10 of the 20 amino acids). Obtain the relaxed protein structure following each mutation from Rosetta and compare the energy of the resulting structures. Which mutant is most stabilizing and which is most destabilizing? Why do you think this is the case?
Documentation:
We installed Rosetta 2017.08 on a macOS machine. To calculate the difference of energy after introducing point mutations, we modified the calculate protein protein ddg demo as necessary to work on the 4BMB PDB file. The main steps are: (1) Relaxing the protein PDB file, (2) Repacking the structure, (3) Calculating Delta G of the wild type protein, (4) Repacking the structure with a point mutation at position 19, (5) Calculating Delta G’ of the mutated protein, (6) Calculating the change in binding energy.
To execute (1), we used the following command:
relax.default.macosclangrelease -s 4bmb.pdb -ignore_unrecognized_res -out:path:pdb results -out:file:scorefile results/relax_4bmb.sc -nstruct 1
We renamed the resulting relaxed PDB file to 4bmb_relaxed.pdb. To execute steps (2)-(5), we used the following command:
rosetta_scripts.default.macosclangrelease -parser:protocol mutation_script.xml -s 4bmb_relaxed.pdb -ignore_unrecognized_res -out:path:pdb results -out:path:score results -nstruct 1
Delta G of each of the wild and mutant type proteins appear at the end of the final PDB file. To perform step (6) and calculate the change in binding energe, we use the following command:
| *tail -n 3 4bmb_relaxed_M.pdb | awk ‘BEGIN{i=0}{if($2 != “”) {a[i] = $2; i++}}END{print a[0]-a[1]}’* |
The command above is an example that corresponds to the PDB file after introducing point mutation M at point 19.
Please not that samples files necessary to reproduce results are submitted to the repository (in Pipeline directory). Files correspond to point mutation M at position 19. Namely, these files are 4bmb_relaxed.pdb, 4bmb-mutM.resfile, and mutation_script. Sample output file, 4bmb_relaxed_M.pdb, is also submitted. Wild type PDB file of 4BMB can be downloaded from RCSB Protein Data Bank here.
To rerun all commands successfully, the most recent version of Rosetta as of May 8, 2017 (Rosetta 2017.08) should be used. Rosetta undergoes weekly releases and demos, movers, and protocols are often subject to change.
In addition to running the pipeline to introduce 10 point mutations at position 19 in protein 4BMB, we generated the results with 19 different point mutations (all possible AA single point substitutions) in another protein, namely 1ZXB.
Results:
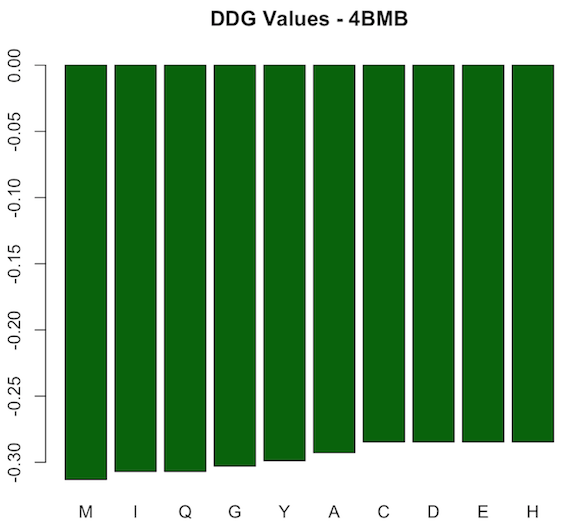
For protein 4BMB, the difference in binding energy (ddG) after introducing the mutation is always negative. However, values vary as shown in the first figure below. The lowest value corresponds to M mutation (-0.31306) and highest to each of C, D, E, and H (-0.28465). Accordingly, all of the mutations are stabilizing ones. The most stabilizing corresponds to M, and the most destabilizing, or in fact the least stabilizing, corresponds to each of the five amino acids C, D, E, and H. The definition of stabilizing (ddG<0) and destabilizing (ddG>0) in terms of the difference in binding energy can be found in paper [1].
According to the FASTA sequence of 4BMB downloaded from its PDB record, the amino acid at position 19 in the wild type protein is T, which is a very hydrophobic acid at pH = 2 and pH = 7 according to lectures by Professor O’Hern (slide 7, lecture 1). The most stabilizing mutation corresponds to M, which is also in the very same category and is the closest to T. That would be a significant reason behind M being the most stabilizing mutation, as both T and M are very hydrophobic and have similar chemical structure. Both amino acids tend to be very repelled from water, are composed mostly of carbon and hydrogen, and have very small dipole moments [2]. Other amino acids mentioned above are also hydrophobic, but their chemical structure is not as close to T’s and accordingly are classified as “hydrophobic” instead of “very hydrophobic.” Furthermore, in paper [1], authors suggest that this kind of mutation might be responsible to not only changing the stability of a protein, but also changing its function.
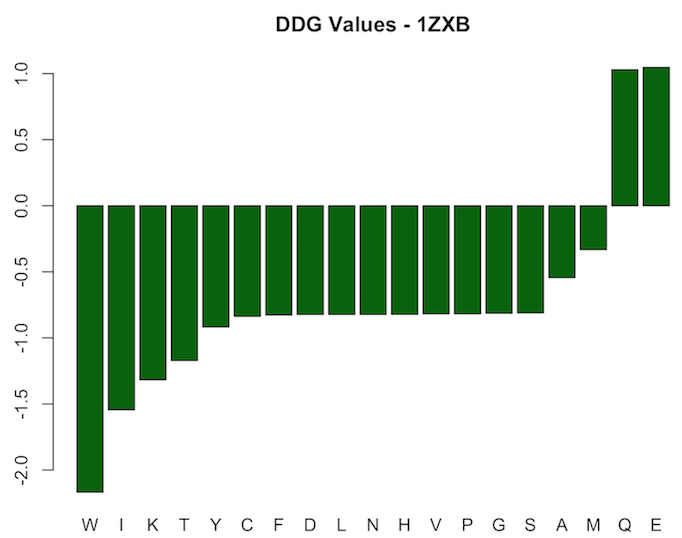
For protein 1ZXB, results resemble a different pattern. Most mutations at point 19 lead to negative change in binding energy, i.e. were stabilizing mutations. However, the two mutations corresponding to Q and E are destabilizing ones and lead to positive one. Values range from -2.1682 to 1.0467. Results are shown in the figure below.
Conclusions:
- Replacing a “very hydrophobic” amino acid with another might lead to the highest stabilizing effect in 4BMB
- Point mutations at position 19 (for 10 different amino acids) in 4BMB lead to further stabilization of the protein
- Point mutations in proteins can lead to either stabilization or destabilization of the structure
- Changes in stability resulting from point mutations might lead to changes in (or loss of) a protein’s function [1]
Source code
References:
[1] Y. Bromberg and B. Rost (2009). Correlating protein function and stability through the analysis of single amino acid substitutions. BMC Bioinformatics, 10(S8).
[2] Biomolecules: Protein 1, University of Wisconsin-Madison. Retrieved on May 8, 2017 from: https://www.chem.wisc.edu/deptfiles/genchem/netorial/modules/biomolecules/modules/protein1/prot13.htm
[3] Yoshida, H., Yamashita, S., Teraoka, M., Itoh, A., Nakakita, S.-i., Nishi, N. and Kamitori, S. (2012), X-ray structure of a protease-resistant mutant form of human galectin-8 with two carbohydrate recognition domains. FEBS J, 279: 3937–3951. doi:10.1111/j.1742-4658.2012.08753.x