Final project 1.3 Final Project
Project Title
Table of Contents
Introduction:
Functional implications of SNP
This project aims at predicting the deleteriousness of SNVs in Carl’s genome. Single nucleotide variants in the coding region of the DNA sequence are likely to affect the structure of the protein product and lead to observable impacts on physiological functioning and disease propensity. Coding region mutations could be classified as silent, missense, nonsense or frameshift mutations based upon the changes in codons and open reading frame. However, deleteriousness could vary significantly among mutations of the same category, and an applicable method to assess deleteriousness must rely upon a more comprehensive prioritization scheme. The standard for determining mutation deleteriousness could either be based upon biochemical or evolutionary information. Bioinformatics tools such as VIPUR incorporates both structural and evolutionary information as the training dataset of the program (Baugh et al., 2016). This strategy allows better analysis of rare proteins with seldom any homology while avoid the oversimplified assumptions of a purely structural approach. However, due to the limited scope for this project, we’ve chosen a first-principle approach that doesn’t involve extensive alignment with homologues.
Writing:
Design of our codes
In the current design of our software, a deleteriousness score between 0 and 1 is calculated for each SNV tested. SNVs from input data are divided into three major categories: 1. indels and introduction or disruption of stop codons; 2. single amino acid substitution and 3. synonymous mutations. Initial deleteriousness scores of 0.8, 0.01 and 0 are assigned for each category, respectively. Frameshift and alteration in stop codons that introduces the addition, deletion or substitution of more than 5 amino acids from the ORF will have an additional 0.20 increase in deleteriousness score. Otherwise, change in each amino acid will yield a 0.04 increase in deleteriousness.
Deleteriousness score for the substitution of single amino acid will be determined by calculating deviance of the substituted amino acid in hydrophobicity index, charge, percent of buried residues, accessible surface area, solubility and volume from the original amino acid residue. Evolutionary constraint upon single amino acid substitution derived from Genomic Evolutionary Rate Profiling is incorporated as an additional dimension (Cooper et al., 2005). In order to weight the different properties, linear regression is performed to identify the significant predictors.
For synonymous mutations, a minor score is added to the output by calculating the changes in codon bias from the codon usage bias table. New codons more favored will not change the value of the output; for the introduction of codons less favored than the original one, the addition in deleterious score is calculated as 0.01x (fraction of original codon usage – fraction of new codon usage) / (fraction of original codon usage).
Additional information not included
Single amino acid conformation vs. chemical environment
The distribution of amino acid psi and phi angles has a significant impact upon the local conformation of peptide chain. Introduction of amino acids that has unusual distribution of phi and psi angles (for example proline or glycine) is likely to disrupt local secondary structure and yield functionally defective proteins. However, the impact of altered backbone conformation may vary according to the chemical environment of a particular residue. The same principle also applies for changes in side chain hydrophobicity and bulkiness Residues that are buried in the interior of a protein fold and essential for protein folding might be more sensitive to changes in side chain size and hydrophobicity, while the linker peptide between folds would adopt a more relaxed conformation and less susceptible to changes in folding and function upon single amino acid substitution. Predicting protein domains and folds is aided by computational comparison across homologues and molecular modeling (Cooper and Shendure, 2011); however, processing large amount of SNVs in structural simulation is still a computationally demanding task.
Active and modification sites
Amino acid residues at enzyme active and crucial modification sites are highly conserved; thus evolutionary information is essential in the detection of deleterious mutations in these regions. However, certain post-translational modifications (ubiquitination, O-GlcNAcylation) utilize multiple sites on the target protein to fine-tune the protein function (Bond and Hanover, 2015). Assessing the deleteriousness of mutations at these modification sites could be challenging at the absence of relevant experimental data. In addition, rules governing the binding of those modification enzymes are less well characterized. Mutations on protein surface that potentially hinders interaction with modification enzymes add yet another layer of complexity to the problem.
Haploinsufficiency and Dosage effect
Nonsense mutations could be either innocuous or harmful depending upon the haploinsufficiency of the gene. It should be noted that the distinction between haplosufficient and haploinsufficient genes might not be absolute; the functional impact of gene dosage could vary across a wide spectrum. Predicting haploinsufficiency relies upon relevant experimental and clinical data; on the other hand, assessing the impact of a particular mutation upon the “effective dosage” of a protein-encoded gene requires additional bioinformatics resources.
Isoforms and splicing variants
Alternative splicing is essential for cellular function especially in tissues such as the CNS, and mutations that affect splicing patterns have been linked to genetic diseases(Wang and Cooper, 2007). SNVs could alter splicing pattern through 1. directly disrupting 5’ and 3’ splice sites; 2. activating cryptic splice site; 3. affect binding of splicing factors to ESE, ESS, ISE and ISS. Mutations in intronic splicing enhancers or silencers, despite located in non-coding regions, could significantly alter the sequences of product peptides. Incorporating splice site prediction could thus provide useful information to SNV deleteriousness assessment.
The deleteriousness of mutations specific to a few isoforms could be challenging to assess if the physiological functions of different isoforms remain largely unknown. Computationally, estimating the functional impact of such mutations is intrinsically defective without supporting data from experiments.
Reference:
Baugh, E.H., Simmons-Edler, R., Müller, C.L., Alford, R.F., Volfovsky, N., Lash, A.E., and Bonneau, R. (2016). Robust classification of protein variation using structural modelling and large-scale data integration. Nucleic Acids Research 44, 2501-2513.
Bond, M.R., and Hanover, J.A. (2015). A little sugar goes a long way: The cell biology of O-GlcNAc. The Journal of Cell Biology 208, 869.
Cooper, G.M., and Shendure, J. (2011). Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet 12, 628-640.
Cooper, G.M., Stone, E.A., Asimenos, G., Green, E.D., Batzoglou, S., and Sidow, A. (2005). Distribution and intensity of constraint in mammalian genomic sequence. Genome Research 15, 901-913.
Wang, G.-S., and Cooper, T.A. (2007). Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 8, 749-761.
Coding:
Documentation:
Introduction
Deleteriousness Estimation V2 is a tool used for estimating the deleteriousness of a mutation based on properties of amino acids and evolutionary constraint.
Usage
python3 deleteriouness_estimation_v2.py -i <input file>
Example
python3 deleteriouness_estimation_v2.py -i input.txt
Input File
Z.3DStruct_annotation.txt
Example Line
variant: ENSG00000005421.4 chr7:94946084:A:T GERP=-1.76;PON1 nonsynonymous ENST00000222381.3 1068_163_55_L->M MAKLIALTLLGMGLALFRNHQSSYQTRLNALREVQPVELPNCNLVKGIETGSEDLEILPNGLAFISSGLKYPGIKSFNPNSPGKILLMDLNEEDPTVLELGITGSKFDVSSFNPHGISTFTDEDNAMYLLVVNHPDAKSTVELFKFQEEEKSLLHLKTIRHKLLPNLNDIVAVGPEHFYGTNDHYFLDPYLQSWEMYLGLAWSYVVYYSPSEVRVVAEGFDFANGINISPDGKYVYIAELLAHKIHVYEKHANWTLTPLKSLDFNTLVDNISVDPETGDLWVGCHPNGMKIFFYDSENPPASEVLRIQNILTEEPKVTQVYAENGTVLQGSTVASVYKGKLLIGTVFHKALYCEL MAKLIALTLLGMGLALFRNHQSSYQTRLNALREVQPVELPNCNLVKGIETGSEDMEILPNGLAFISSGLKYPGIKSFNPNSPGKILLMDLNEEDPTVLELGITGSKFDVSSFNPHGISTFTDEDNAMYLLVVNHPDAKSTVELFKFQEEEKSLLHLKTIRHKLLPNLNDIVAVGPEHFYGTNDHYFLDPYLQSWEMYLGLAWSYVVYYSPSEVRVVAEGFDFANGINISPDGKYVYIAELLAHKIHVYEKHANWTLTPLKSLDFNTLVDNISVDPETGDLWVGCHPNGMKIFFYDSENPPASEVLRIQNILTEEPKVTQVYAENGTVLQGSTVASVYKGKLLIGTVFHKALYCEL
Other Files Required
Other files required include PAM250.txt, codon_usage.txt and scaled_aa_info2.txt. All these files should be downloaded in the working directory.
Output Format
Mutation_id Gene Mutation_type Estimation_score
Results:
- The score table is included in the repo. Score Table
- The score distribution.
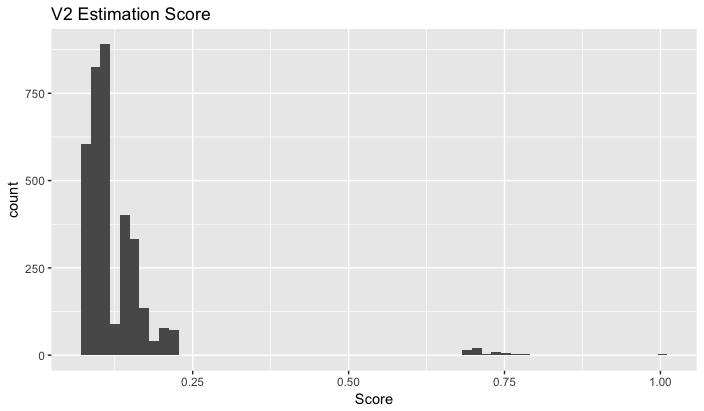
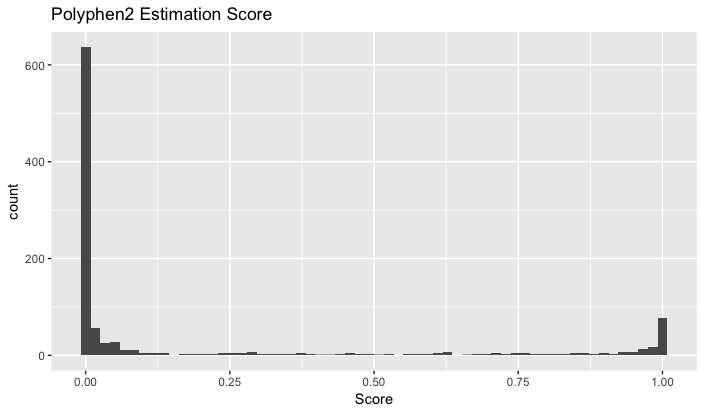
Pipeline:
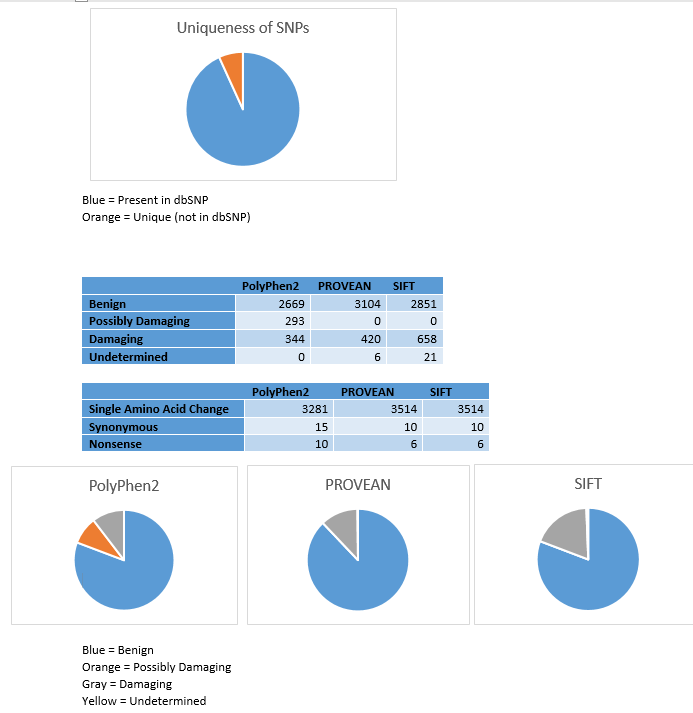
Many SNPs are quite common within a population. The output of PROVEAN, one of the programs used to analyze the deleteriousness of a particular variation, provides information on whether the individual SNPs are found within the SNP database or if they are a unique variant. Of the 3530 protein coding SNPs analyzed, 93.2% (3290) were present in the dbSNP, while only 6.8% (240) were unique mutations.
Some of the SNPs present in the VCF file are isoforms of the same protein. In the case where each isoform has the same mutation, it will likely have the same effect and classification (due to the similarity between the proteins). However, some cases may have different mutations in each isoform, with different classifications for the different variations. In either case, the presence of non-mutated isoforms or isoforms with benign mutations could have the ability to compensate for any function lost by a damaging variant. This complicates deleteriousness predictions, as it is possible that mutations classified as harmful, which likely have greatly altered structure and function, may not actually be deleterious overall, as the isoform with a proper structure can carry out the required functions.
The program PolyPhen2 was used to analyze Carl’s SNPs. This program calculates the probability that a particular SNP is “damaging”, using a Bayes classifier and information about the particular sequence, its conservation, and the structure of the gene (Adzhubei et al. 2013). The only input required is the chromosome number, the location of the gene within the chromosome, and the original and variant nucleotide. 3306 of the 3533 protein coding SNPs were analyzed using this method, with 89 of the original SNPs classified as belonging to introns, 17 to the 3’ UTR, and 11 to the 5’ UTR. Of the protein coding variants, 80.7% (2669) were classified as “benign”, with probabilities of being damaging ranging from 0 to .451. 10.4% (344) of the SNPs were predicted to be “probably damaging”, with a probability ranging from .959 to 1. SNPs were also classified as “possibly damaging”; 8.9% (293) of these fell into this category, with probabilities ranging from .454 to .948.
A ranking of the deleteriousness of the different SNPs was also created using PolyPhen2. The ranking is from a PolyPhen score of 1 (a very probable damaging mutation) to 0 (indicating a benign mutation). This can be compared to the ranking obtained through the other programs or through the program created, and can asses how closely these programs can classify and rank the different SNPs.
The algorithm PROVEAN, assigns the SNPs as either “neutral” or “deleterious” through alignment base scoring (Choi and Chan, 2015). Of the 3533 SNPs input into the program, 3 were determined to be in a non-protein coding region, and were not analyzed further. The remaining 3530 protein coding SNPs were analyzed using this program. 87.9% (3104) were classified as neutral, with 11.9% (420) classified as deleterious. A further 0.2% (6) were unable to be classified. Of the neutral variations, 99.7% were single amino acid changes, while 0.3% were synonymous mutations. All of the deleterious mutations involved single amino acid changes, while all of the undetermined variants encoded nonsense mutations.
The program SIFT was also used to analyze the 3530 protein coding SNPs (3 of the original 3533 were also found to be in non-protein coding regions). It uses sequence alignment and conservation to determine the probability of a particular mutation occurring, classifying a mutation as “damaging” if this value is below a particular cutoff (Kumar et al. 2009). 80.8% (2851) were considered to be “tolerated”, while 18.6% (658) were considered to be “damaging”. A further 0.6% (21) were undetermined. This program also gave an identical prediction with regards to the nonsynonymous and synonymous classification of the tolerated and damaging SNPs. Likewise, 6 variants classified as “undetermined” were considered to be nonsense mutations, the exact number as was found using PROVEAN.
Each of the programs was fairly consistent in their classification of a particular SNP as benign or neutral. PolyPhen2 and SIFT had nearly identical classification into this category (80.8% vs 80.7%), while PROVEAN predicted slightly more neutral variants (87.9%). The three-category classification used in PolyPhen2 meant that fewer variants were predicted to be damaging, with 10.4% compared to the 11.9% and 18.6% seen with PROVEAN and SIFT respectively. Due to the very similar probabilities, SIFT likely classifies the “possibly damaging” mutants seen with PolyPhen2 as “damaging”.
Both PROVEAN and SIFT identified the same number of nonsynonymous, synonymous, and nonsense mutations. This implies that these algorithms can correctly classify the type of mutation, and that both programs have the ability to correctly recognize the codons within the genes and whether the mutation affects the amino acid being encoded. PolyPhen2 has similar results, with slightly fewer synonymous mutations and nonsense mutations detected. The similarity between these programs with regards to codon detection and deleteriousness predictions suggests that though the different algorithms classify the majority of SNPs in a similar way, there are still some variants which are variably classified. Experimental analysis through mutagenesis will be helpful in determining whether these variably classified SNPs are truly harmful or benign.
Source code
References:
Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Current protocols in human genetics / editorial board, Jonathan L Haines . [et al]. 2013;0 7:Unit7.20.
Choi Y., Chan A.P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745-2747.
Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4(8):1073-1082.